





文章已经过时,请去官网查阅相关文档
摘要
一文介绍单细胞测序生物信息分析完整流程,这可能是最新也是最全的流程
基础流程(cellranger)
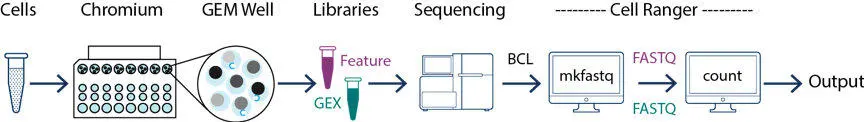
cellranger 数据拆分
cellranger mkfastq 可用于将单细胞测序获得的 BCL 文件拆分为可以识别的 fastq 测序数据
cellranger makefastq --run=[ ] --samplesheet=[sample.csv] --jobmode=local --localcores=20 --localmem=80 |
-–run :是下机数据 BCL 所在的路径;
-–samplesheet :样品信息列表–共三列(lane id ,sample name ,index name)
注意要控制好核心数和内存数
运行产出结果存在于 out 目录中
cellranger 数据统计
cellranger count是 cellranger 最主要也是最重要的功能:完成细胞和基因的定量,也就是产生了我们用来做各种分析的基因表达矩阵。
cellranger count \ |
id :产生的结果都在这个文件中,可以取几号样品(如 sample345);
fastqs :由 cellranger mkfastq 产生的 fastqs 文件夹所在的路径;fastqs :由 cellranger mkfastq 产生的 fastqs 文件夹所在的路径;
indices:sample index:SI-3A-A1;
transcriptome:参考转录组文件路径;
cells:预期回复的细胞数;
下游分析
cellranger count 计算的结果只能作为初步观测的结果,如果需要进一步分析聚类细胞,还需要进行下游分析,这里使用官方推荐 R 包(Seurat 3.1)
流程参考官方(外周血分析标准流程)
软件安装
install.packages('Seurat') |
生成 Seruat 对象
library(dplyr) |
## An object of class Seurat |
这里读取的是单细胞 count 结果中的矩阵目录;
在对象生成的过程中,做了初步的过滤;
留下所有在>=3 个细胞中表达的基因 min.cells = 3;
为了除去一些质量差的细胞,留下所有检测到>=200 个基因的细胞 min.genes = 200。
标准预处理流程
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats |
这一步 mit-开头的为线粒体基因,这里将其进行标记并统计其分布频率
# Visualize QC metrics as a violin plot |
对 pbmc 对象做小提琴图,分别为基因数,细胞数和线粒体占比
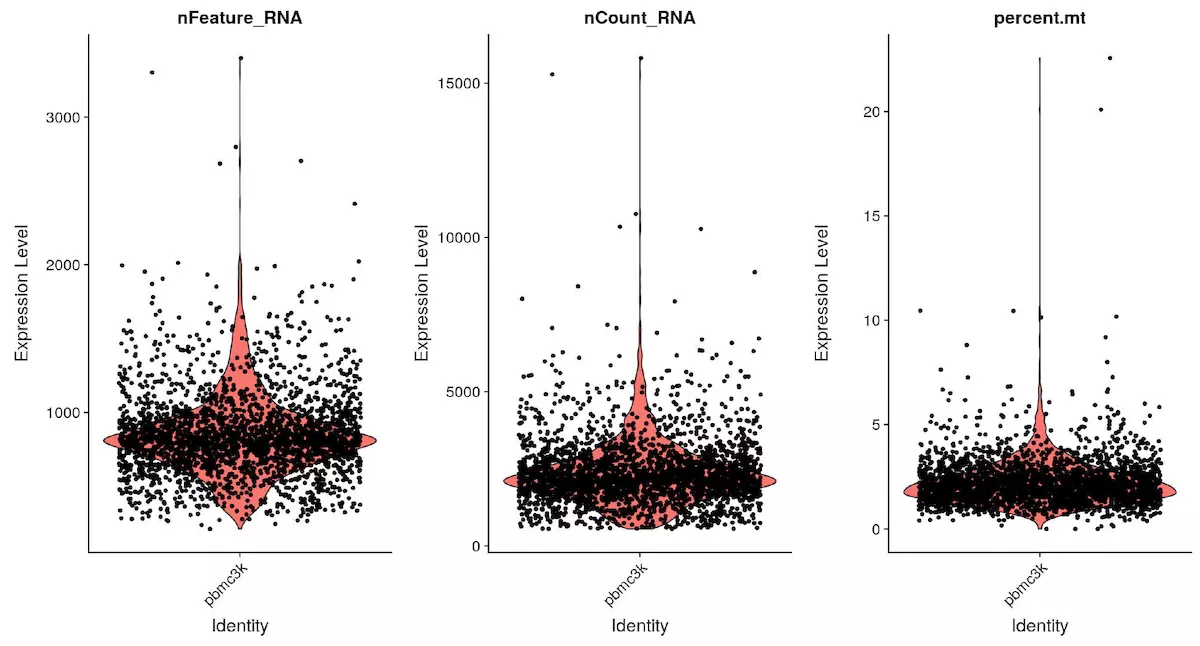
pbmc <- subset(x = pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5) |
接下来,根据图片中基因数和线粒体数,分别设置过滤参数,这里基因数 200-2500,线粒体百分比为小于 5%
数据标准化
pbmc <- NormalizeData(object = pbmc, normalization.method = "LogNormalize", scale.factor = 10000) |
鉴定高度变化基因
pbmc <- FindVariableFeatures(object = pbmc, selection.method = "vst", nfeatures = 2000) |
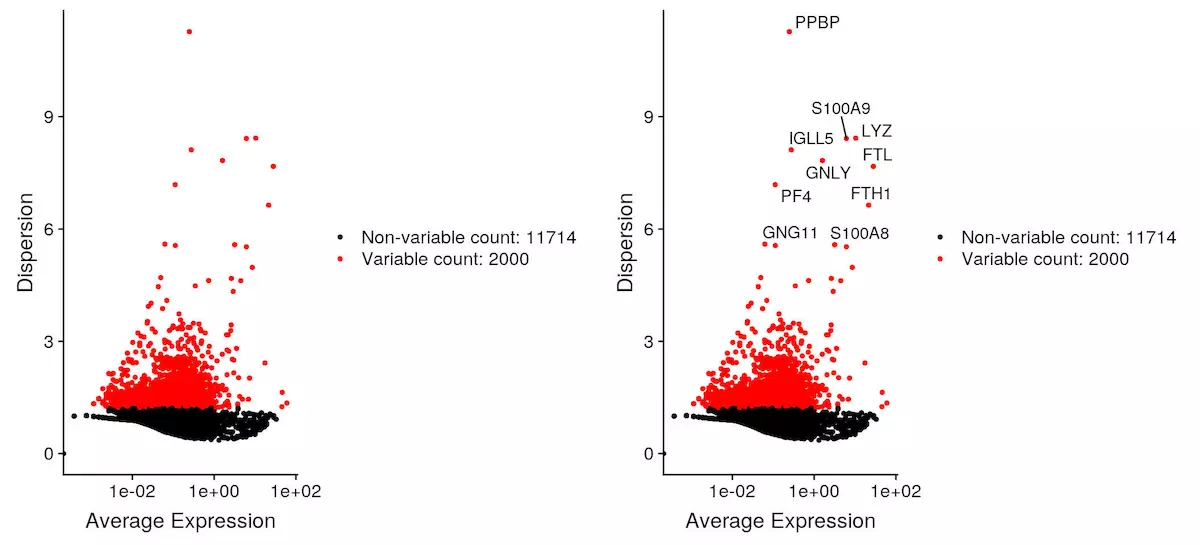
数据归一化
all.genes <- rownames(x = pbmc) |
这里设置对所有的基因都做了scale,但是需要知道的是,其实后续的分析都是基于高变基因的,因此,使用默认参数就可以了,而且提升效率。
pbmc <- ScaleData(object = pbmc) |
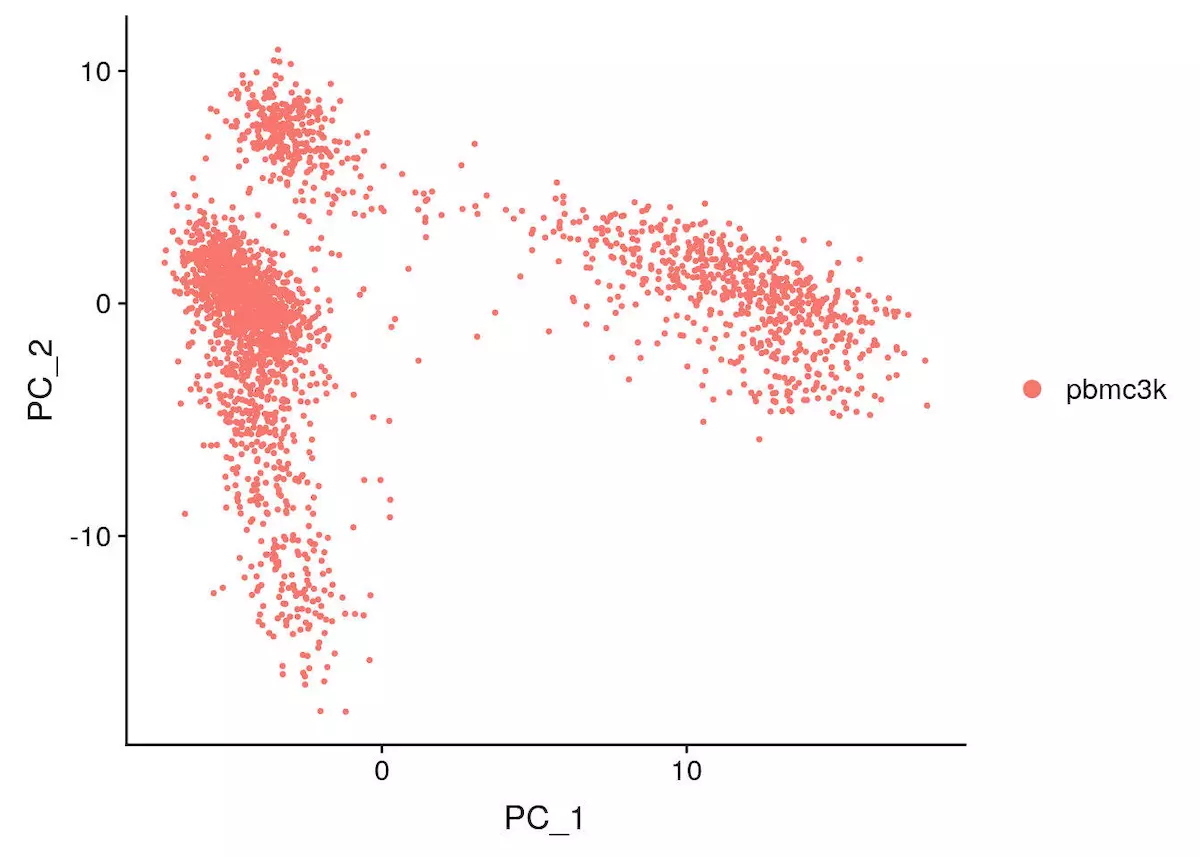
线形降维
pbmc <- RunPCA(object = pbmc, features = VariableFeatures(object = pbmc)) |
这里有多种方法展示 pca 结果,本文采用最简单的方法
DimPlot(object = pbmc, reduction = "pca") |
鉴定数据集的可用维度
pbmc <- JackStraw(object = pbmc, num.replicate = 100) |
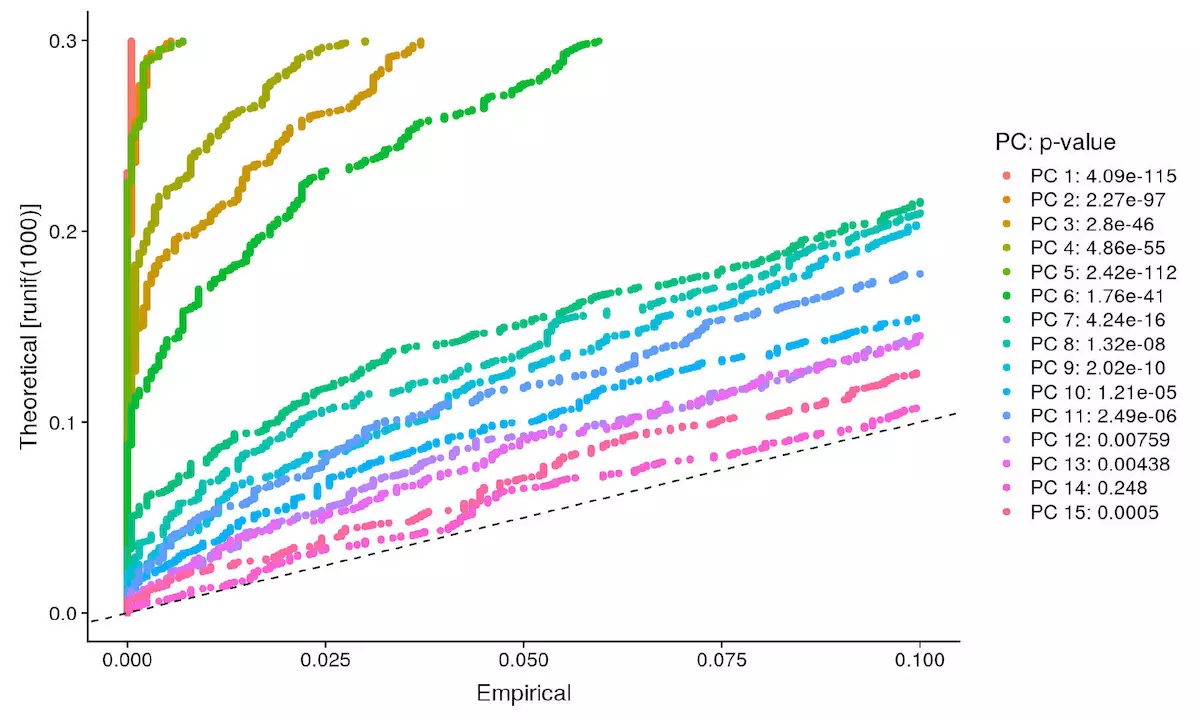
虚线以上的为可用维度,你也可以调整 dims 参数,画出所有 pca 查看
另外一种鉴定手段是绘制所有 PC 的分布点图
ElbowPlot(pbmc) |
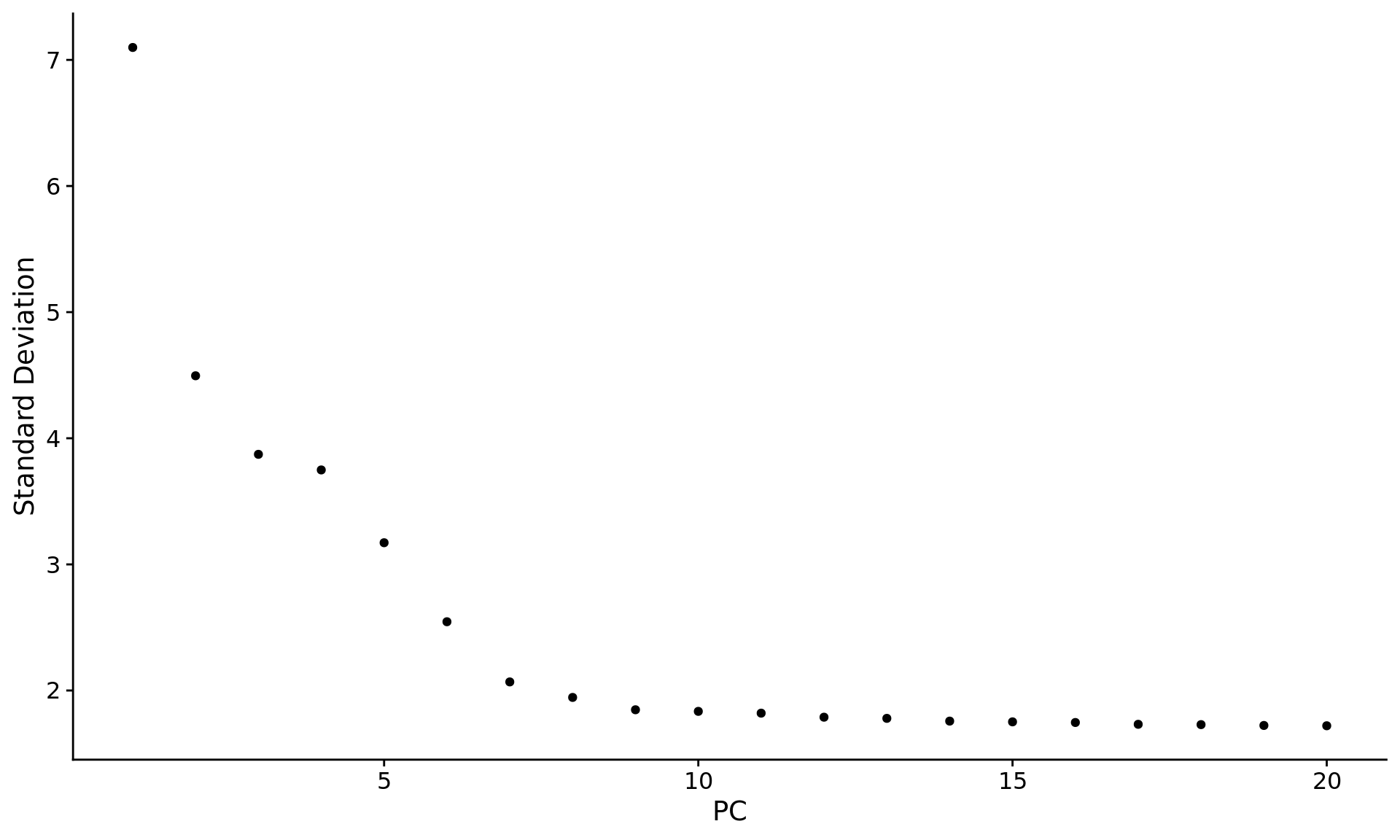
大多数软件都是通过拾取拐点处的 pc 作为选定数目
细胞聚类
pbmc <- FindNeighbors(object = pbmc, dims = 1:10) |
这里的 dims 为上一步计算所用的维度数,而 resolution 参数控制聚类的数目,针对 3K 的细胞数目,最好的范围是0.4-1.2
head(Idents(pbmc), 5) |
## AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC AAACCGTGCTTCCG AAACCGTGTATGCG |
执行非线性降维
这里注意,这一步聚类有两种聚类方法(umap/tSNE),两种方法都可以使用,但不要混用,这样,后面的结算结果会将先前的聚类覆盖掉,只能保留一个
本文采用基于 umap 的聚类方法
pbmc <- RunUMAP(object = pbmc, dims = 1:10) |
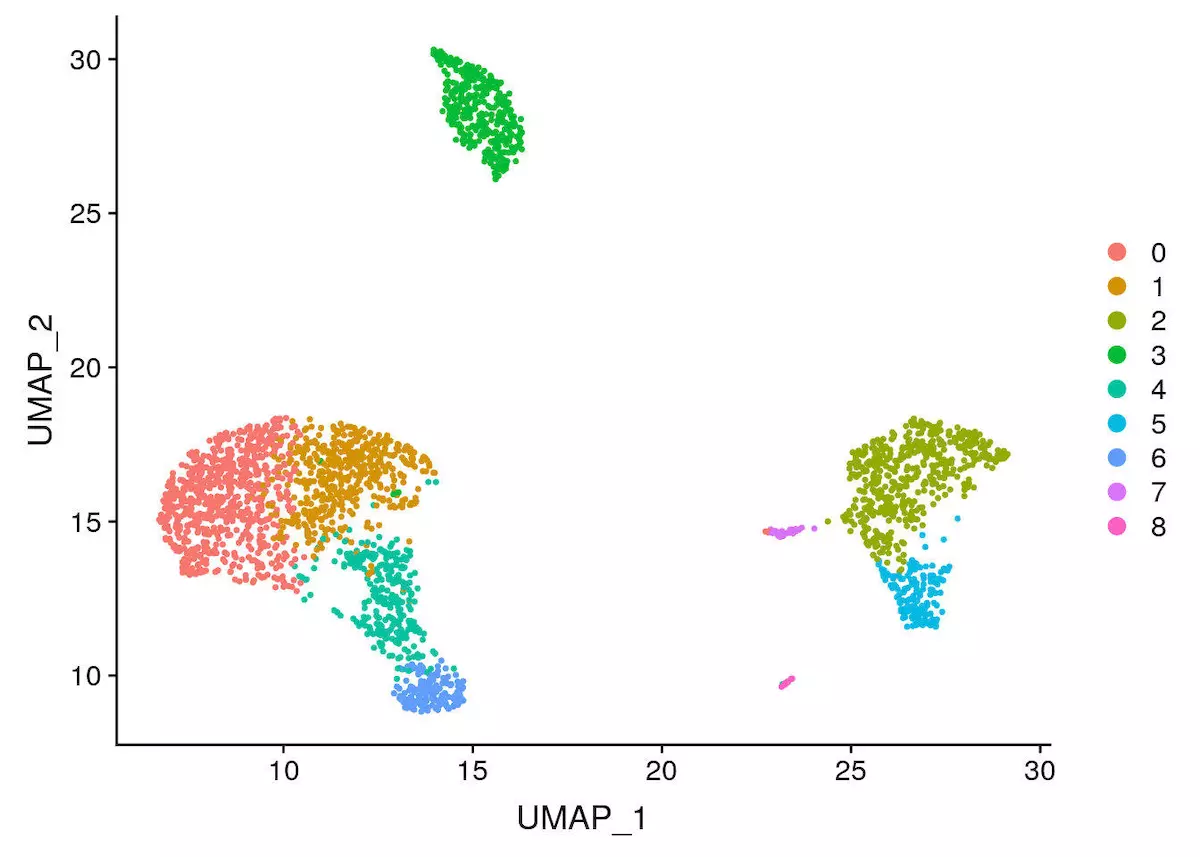
完成聚类后,一定要记住保存数据,不然重新计算可要头疼了
saveRDS(pbmc, file = "../output/pbmc_tutorial.rds") |
寻找每个聚类中显著表达的基因
cluster1.markers <- FindMarkers(object = pbmc, ident.1 = 1, min.pct = 0.25) |
这样是寻找单个聚类中的显著基因
cluster5.markers <- FindMarkers(object = pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25) |
这样寻找所有聚类中显著基因,计算速度很慢,需要等待
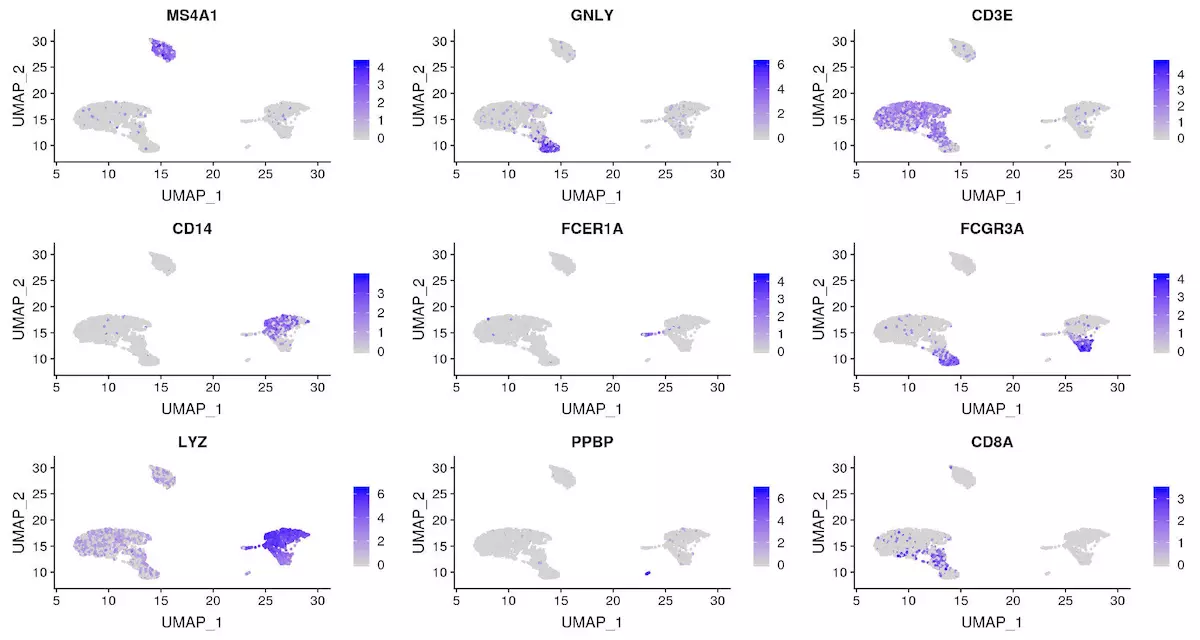
另外,我们有多种方法统计基因的显著性
FeaturePlot(object = pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", |
top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC) |

剩下的便是寻找基因 marker 并对细胞类型进行注释
你可能想要知道如何对多样本进行整合分析,请参考教程:整合刺激性和对照性 PBMC 数据集,以学习细胞类型特异性反应
你可能想了解如何灵活操作 Seurat 的 S4 对象,以便轻松的提取表达矩阵;亦或者想要做一些不一样的可视化效果,例如采用平均表达量做热图展示等,请参考Seurat3.1 的灵活操作指南
你可能想对单细胞数据做进一步分析,例如功能分析,请参考10X 单细胞数据针对细胞及其亚型的基因集功能分析和转录调控分析
全自动细胞类型注释
众所周知,细胞类型的注释是最困难的一步,除非你有很强的对细胞基因的敏感度,不然很难识别细胞类型。
通常识别细胞类型的方法主要有三种
- 根据 Marker 基因,采用CellMarker或者panglaoDB数据库,进行细胞注释,可以采用超几何分布算法来进行精确性验证,方法参考ClusterProfiler:真的不只是富集分析
- 从文献中获取已经验证的 Marker
- 采用一些自动化注释的软件,例如
SingleR,Garnett,celaref等
这里简单介绍下SingleR
SingleR:一个全自动细胞注释的 R 包,用法很简单
软件安装
BiocManager::install("SingleR") |
创建 SingleR 对象
从头预测的方法请参考官方教程
因为我们刚刚从 Seurat 过来的,所以我们应该很想知道 Seurat cluster 的细胞注释结果,因此,对 Seurat 的结果进行注释
我们这里采用两个人类的参考集去做细胞注释
library(Seurat) |
读入Seurat对象转换为SingleCell支持的对象
seurat.obj <- readRDS("../output/pbmc_tutorial.rds") |
采用两个参考集一起进行注释,
Anno <- SingleR(test = test, |
提取需要的细胞分类信息
Anno$cluster <- rownames(Anno) |
你也可以将细胞注释信息重新添加到Seurat对象中去
new.cluster.ids <- fin$labels |
伪时间分析
伪时间分析建议采用 monocle3.0 软件
软件安装
##安装依赖 |
标准分析流
library(Seurat) |
不要直接把meta.data放入pd中去,太多没用的信息了,先清理下
new_pd<-select(pd,nCount_RNA,nFeature_RNA,percent.mt) |
生成monocle的cds对象
cds <- new_cell_data_set(data,cell_metadata = new_pd,gene_metadata = fData) |
运行下游标准流程,无论如何都得跑,是必须的默认流程
cds <- preprocess_cds(cds, num_dim = 30) |
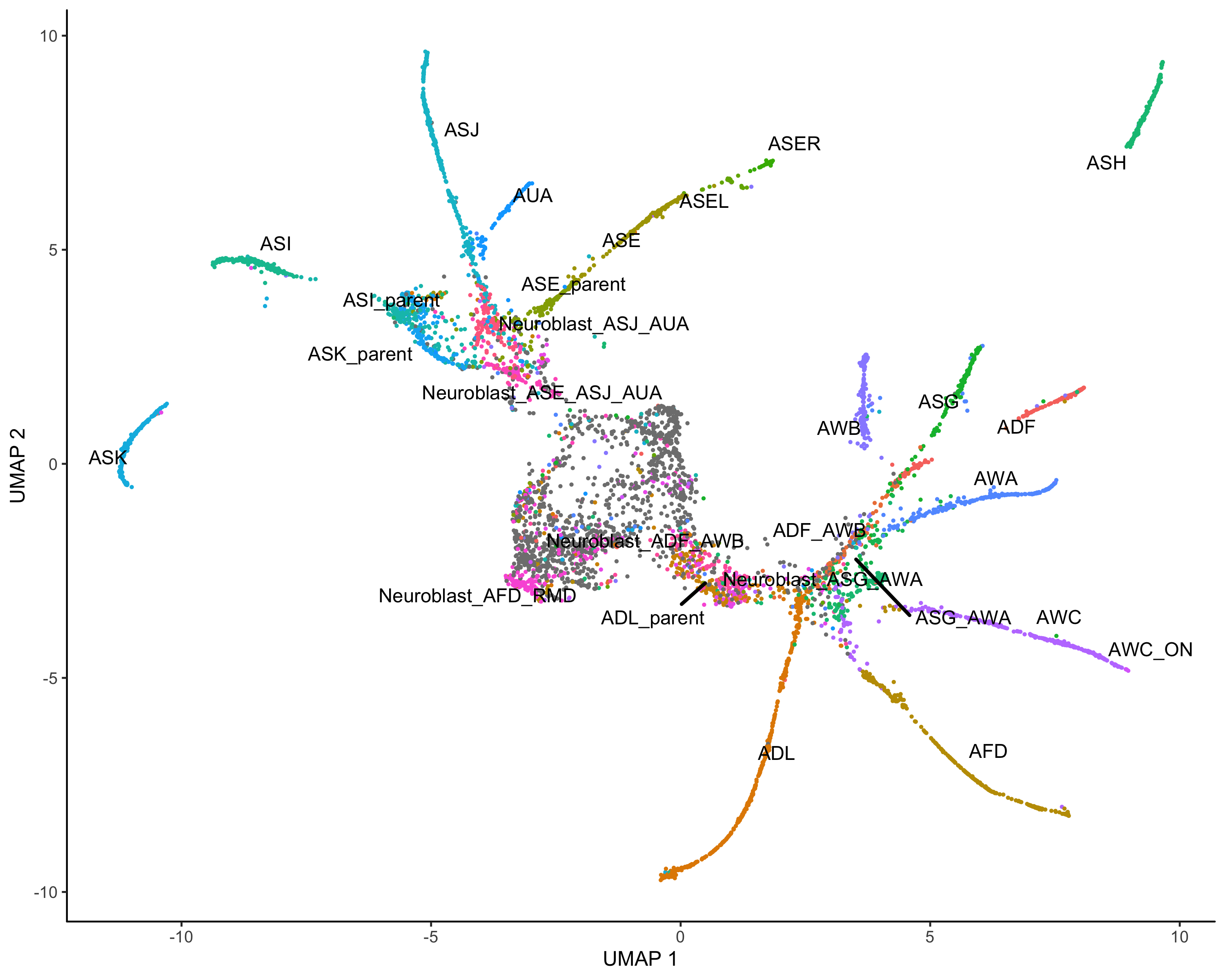
#cluster |
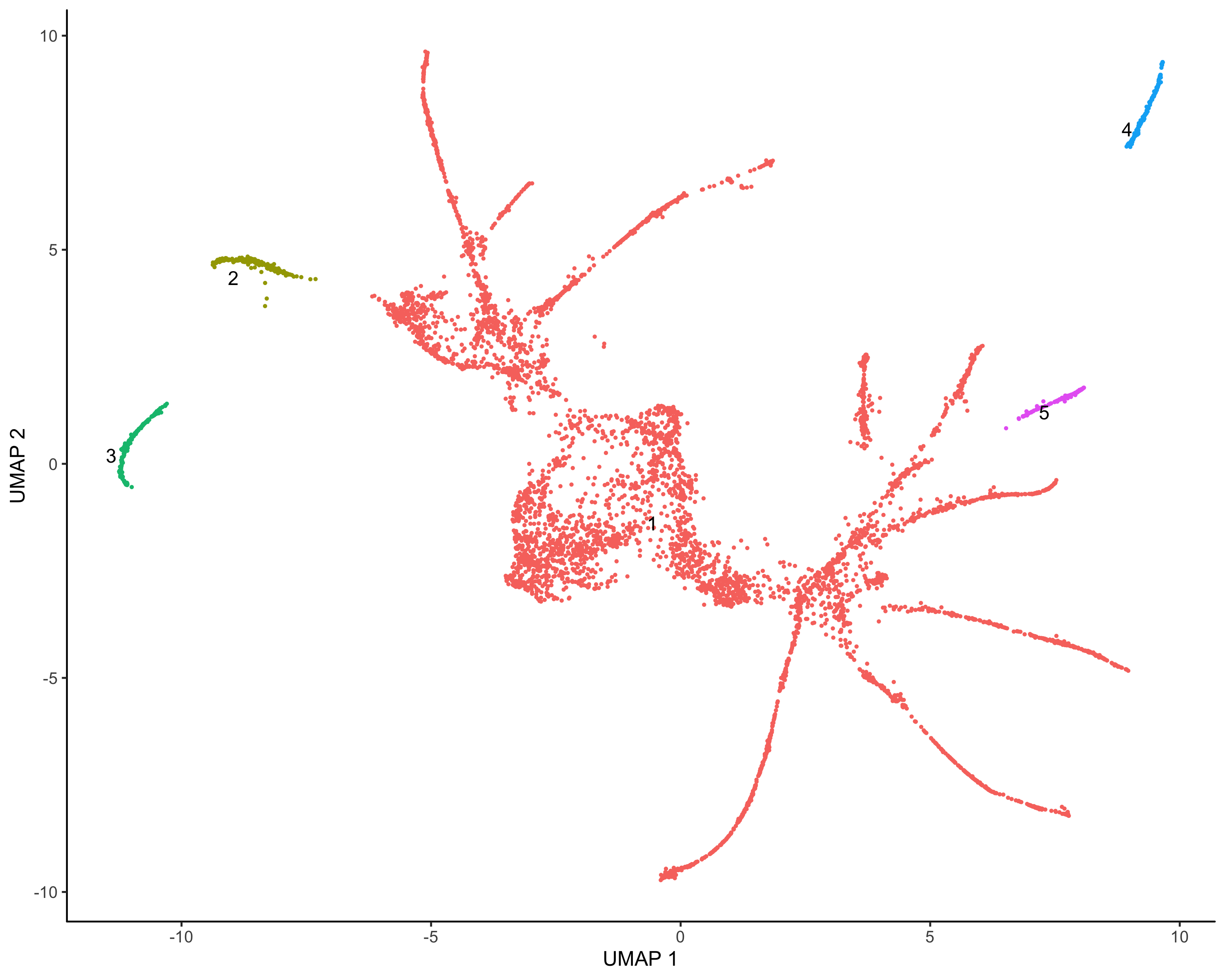
cds <- learn_graph(cds) |
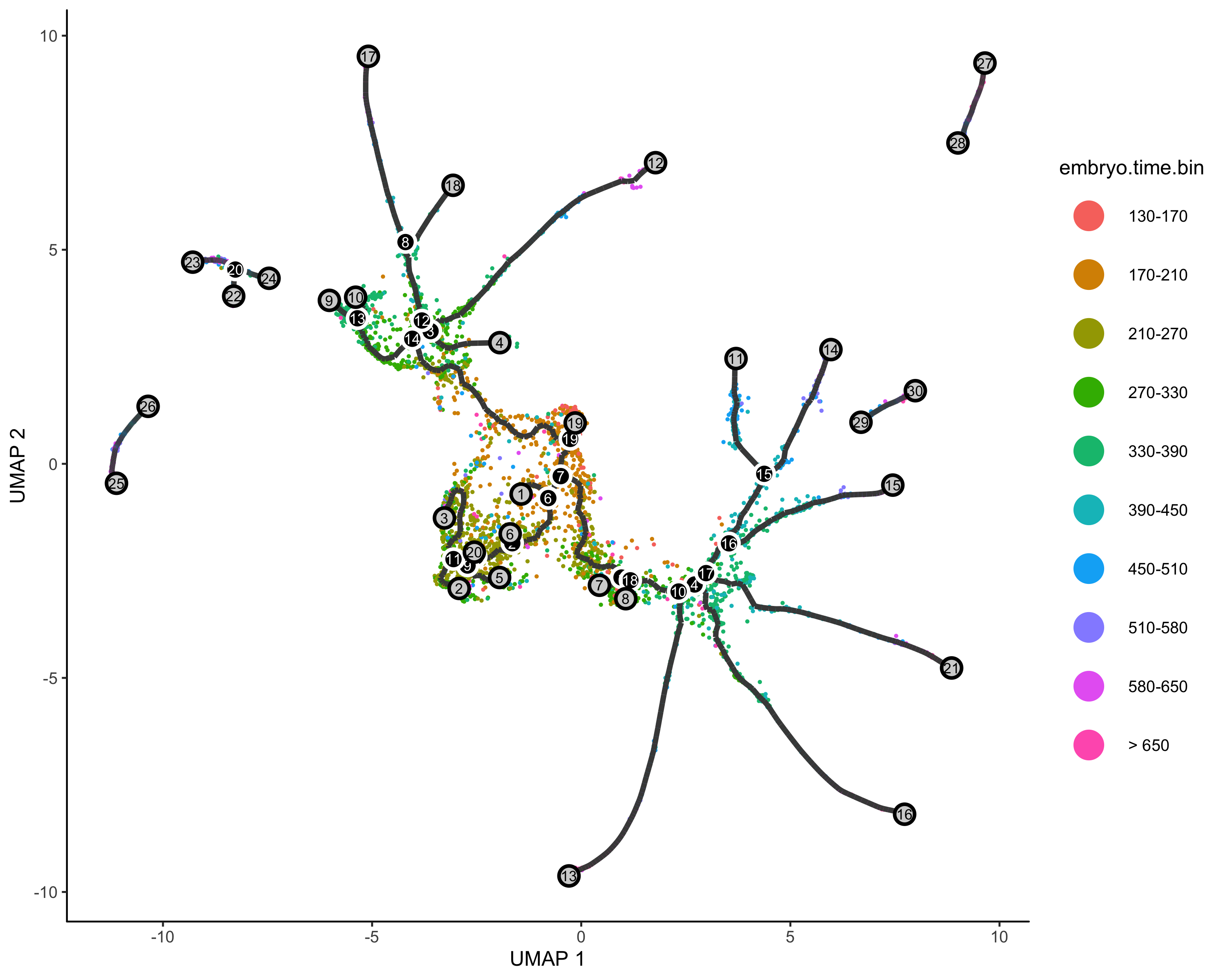
定义时间开始节点&&伪时间分析
##一个有用的寻找起源节点的函数 |
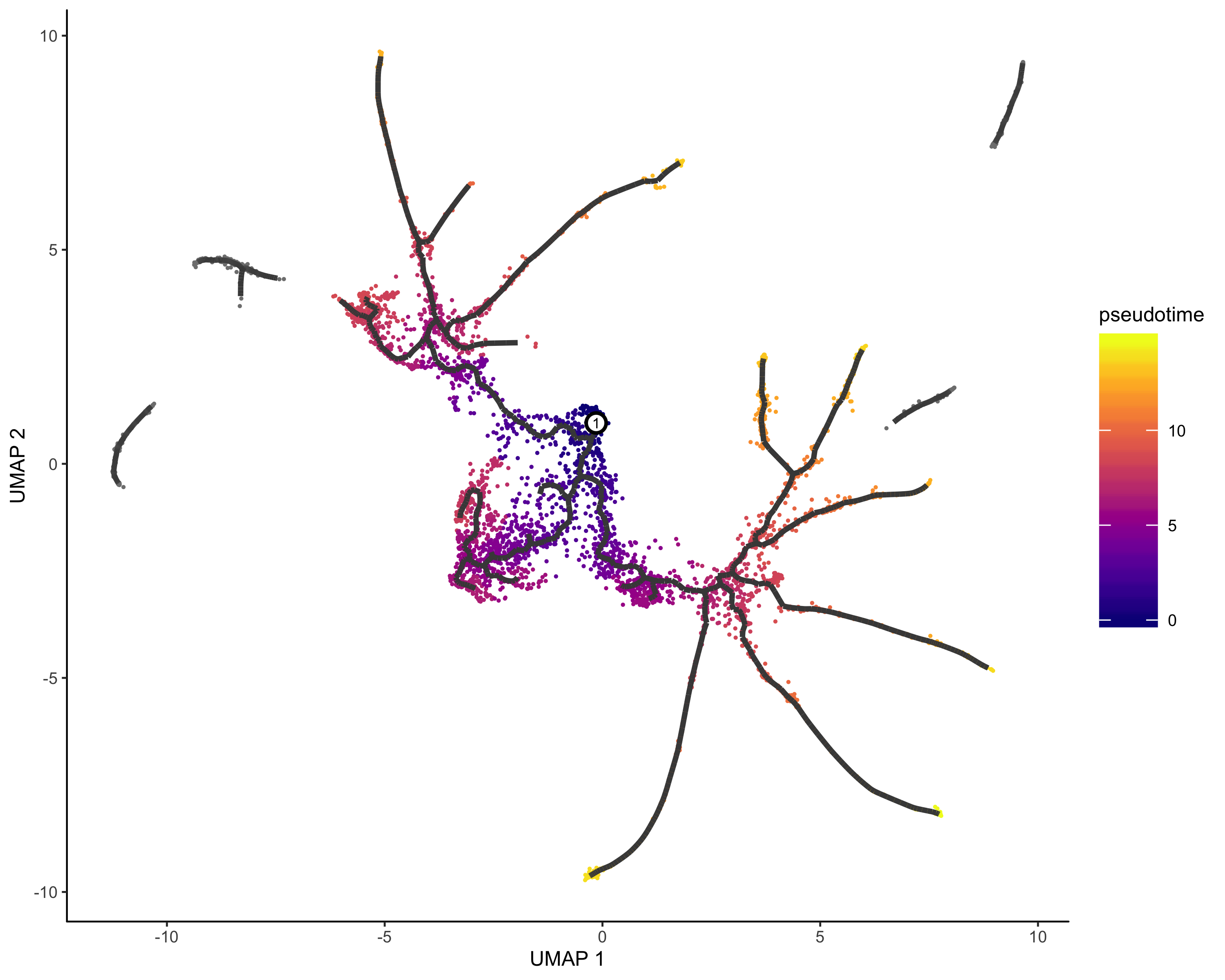
PS:对多样本进行伪时间分析,请参考使用 Monocle3 对多样本单细胞数据进行伪时间分析
本文纯属原创,部分数据采用官方教程,转载需标明出处
教程链接:
