





网上很多教程都在讲Y叔的clusterprofile富集分析的教程,但是查阅了官方文档后才知道,这个包真的不仅仅只有这个功能,其他功能也很强大。
做ID转换
ID转换应该是基因下游分析的敲门砖了,因为一般注释用的是ENSMEBL ID,但是这个ID是人类无法识别的一串数字,下游分析功能都需要转换成基因名称或者基因ID,有这个bitr功能就方便了很多。
x <- c("GPX3", "GLRX", "LBP", "CRYAB", "DEFB1", "HCLS1", "SOD2", "HSPA2", |
## SYMBOL ENTREZID |
想知道你可以进行哪些ID 转换?
library(org.Hs.eg.db) |
## [1] "ACCNUM" "ALIAS" "ENSEMBL" |
Note:虽然GO分析支持很多ID,例如symbol完全可以直接运行,但还是建议都转换为ENTREZID,毕竟,我们很少直接得到symbol啊。
GSEA分析
还在使用官方的GSEA java包进行富集分析吗,你需要准备gct文件(表达矩阵),要转换为symbol或者entrezid,还要准备cls文件(样本特征),还要下载好gmt文件,最后出来的分析结果,各种图片都是不好看的,准确说不够灵活,用R包呢?
GSEA_GO
ego3 <- gseGO(geneList = geneList, |
geneList 是什么?
类似于做富集(过表达)分析,geneList是一列基因id,而GSEA分析一般需要基因表达量来衡量富集分数,但是从原理上来讲,还是根据表达量计算Fordchange,然后给他排序,看这个排序在基因集中的富集程度。这样就不会因为传统分析中先筛选差异基因而过滤掉的低差异基因信息(详情参考GSEA的原理),所以,这个基因列表是指一列排序后的以基因名称为名字的log2FC值向量。
假设你有一个两列的文件,第一列为名字,第二列为logFC,你可以这样:
d <- read.csv(your_csv_file) |
剩余都是一些限制参数,请执行?clusterProfiler::gseGO
## 4312 8318 10874 55143 55388 991 |
GSEA-KEGG
kk2 <- gseKEGG(geneList = geneList, |
## ID Description setSize |
结果里面包含了所有GSEA计算的结果数值
setSize:基因集的大小
enrichmentScore:富集打分
NES:标准化后的富集打分
pvalue,p.ajust.qvalues:各种显著性检验
rank:log2FC的排序位置
根据官方提供的gmt文件或者自己做gmt文件进行分析
假设你从官网下载了Hallmark基因集
wp2gene <- read.gmt("h.all.v7.0.entrez.gmt") |
这样就可以直接得到富集分析的结果,非常方便
另外,也可以通过msigdbr包直接获取基因集信息,但是感觉灵活性不高。
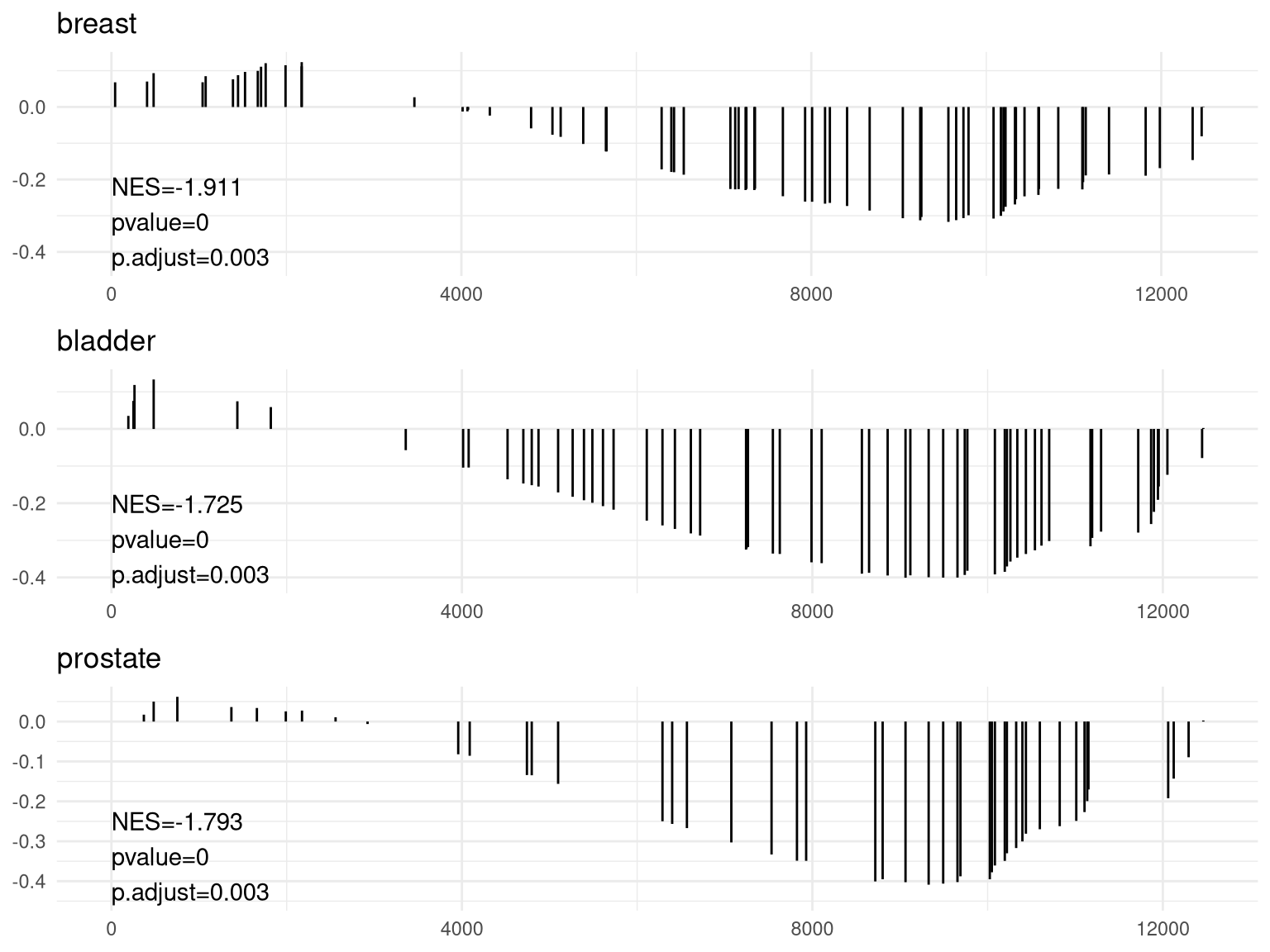
对GSEA的结果可视化
anno <- edo2[1, c("NES", "pvalue", "p.adjust")] |
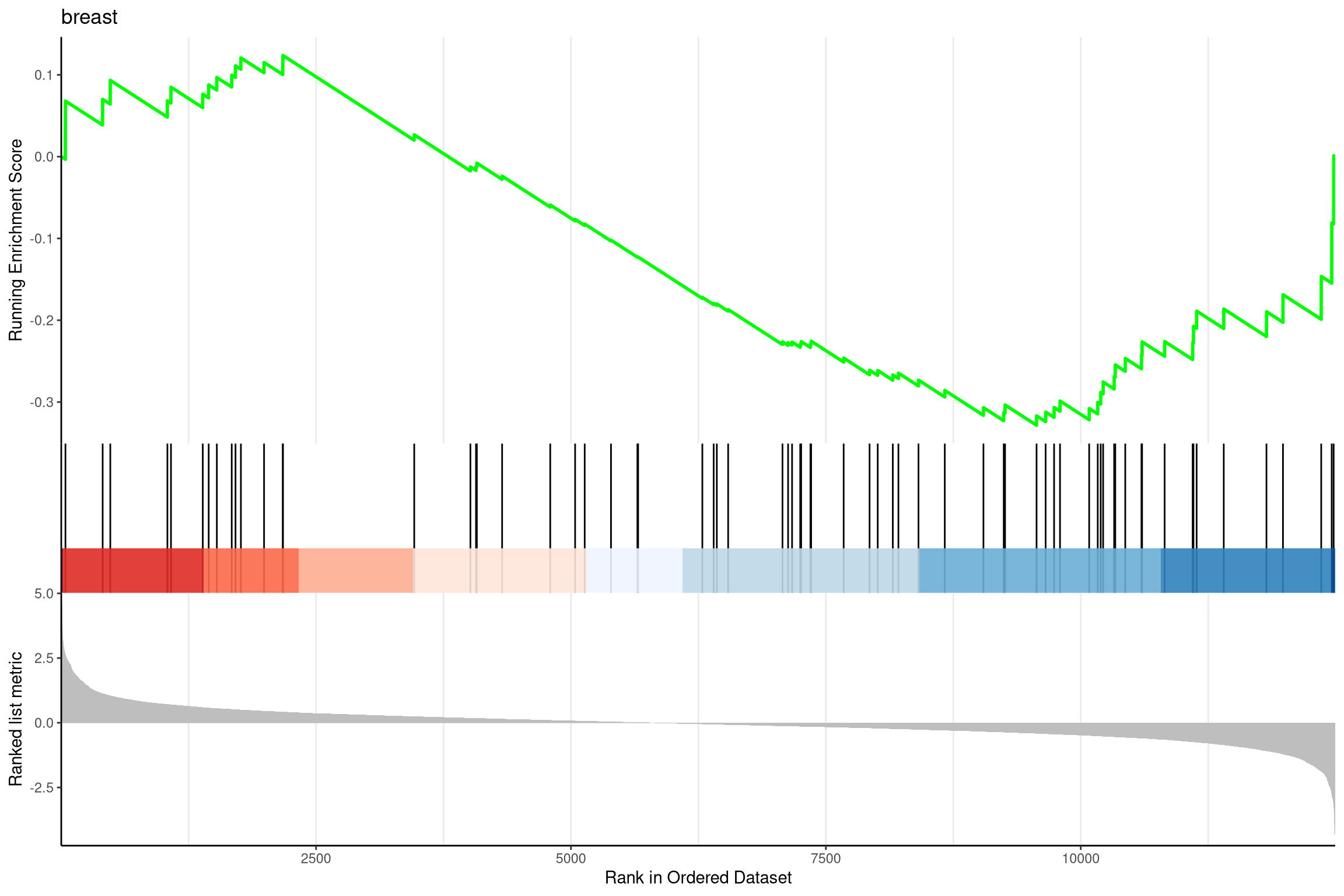
pp <- lapply(1:3, function(i) { |
使结果可读性提升
针对分析结果,GO富集可以设置参数readable = TRUE,但是对KEGG无法使用,因此,可以使用setReadable
library(org.Hs.eg.db) |
## ID Description GeneRatio BgRatio |
y <- setReadable(x, OrgDb = org.Hs.eg.db, keyType="ENTREZID") |
## ID Description GeneRatio BgRatio |
你甚至可以用它来对单细胞聚类结果进行注释
如果我们有一个大的细胞marker库,然后我们有显著差异的基因marker,那寻找细胞类型就和过表达分析是一种情况了,都是采用超几何分布计算概率,所以:
cell_markers <- vroom::vroom('http://bio-bigdata.hrbmu.edu.cn/CellMarker/download/Human_cell_markers.txt') %>% |
你甚至可以从cellmarker官网直接下载所有的细胞marker
## # A tibble: 2,868 x 2 |
y <- enricher(gene, TERM2GENE=cell_markers, minGSSize=1) |
这样就找到了细胞类型,你可以过滤占比最高,且p值显著的结果。
