





本教程中,我们的目标是使用第二代和第三代全基因组测序 (WGS) 数据组装细菌基因组。我们将以此为例来探讨WGS数据分析,并探讨测序技术之间的差异。
软件安装
Docker镜像
您可以使用以下命令从DockerHub中提取镜像:
docker pull yanhui09/mac2023_extra |
用mamba安装
建议使用mamba在独立 conda环境中安装软件。
假设您已经在系统中安装了mamba软件,您可以使用mamba创建一个新conda环境:
转到下载的数据目录。确保您知道自己当前的工作目录位置。
例如,下载的数据目录位于/home/username/MAC2023-extera
cd /home/username/MAC2023-extera |
您将看到下载的数据目录的路径,如下所示。
/home/username/MAC2023-extera |
现在您可以使用以下命令创建新conda环境并安装软件:
mamba env create -n wgs1 -f envs/env1.yaml |
激活环境以进行以下分析。
conda activate wgs1 |
使用演示数据进行探索
WGS 数据可以像以前一样从MAC2023-extra获取。
首先用seqkit来查看数据
seqkit stat data/wgs/*.fastq.gz data/wgs/ncbi_pacbio_TL110.fasta |
预期输出:
file format type num_seqs sum_len min_len avg_len max_len |
这是来自费氏丙酸杆菌菌株的一组测序数据。我们对 Illumina 和 Oxford Nanopore Technologies (ONT) 读数的测序数据进行二次采样,覆盖率达到 20 倍。PacBio 参考基因组来自NCBI RefSeq 数据库。
Q1:该菌株的基因组大小是多少?测序覆盖度是如何计算的?
illumina:30,200,000*2/2,566,312≈20
ONT:51,201,670/2,566,312≈20
我们用fastqc来看看测序数据的质量。
mkdir -p fastqc/illumina fastqc/ont_r10 |
您可以打开.html该fastqc目录下的文件来查看测序数据的质量。
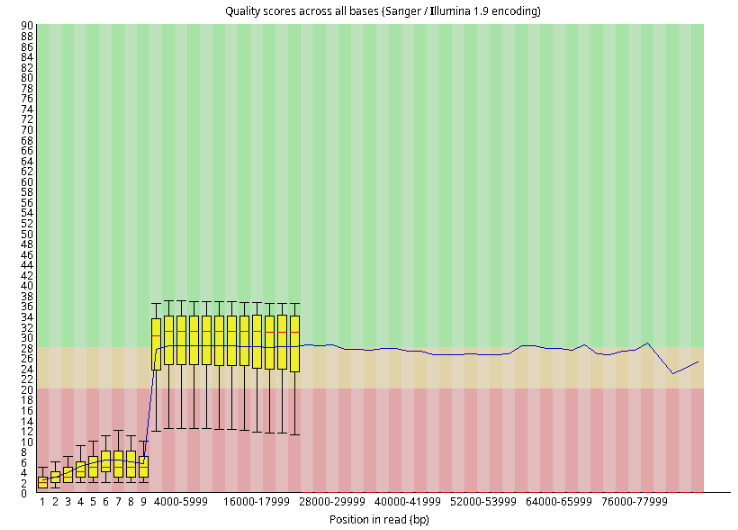
ONT
Illumina
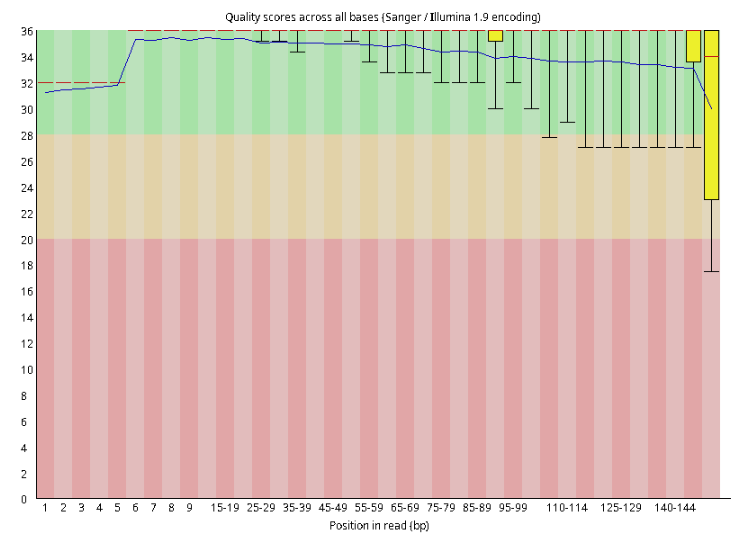
我们可以看到 ONT 读取比 Illumina 读取更长,但包含更多错误。
使用 Illumina 数据进行基因组组装
trimmomatic去除接头
trimmomatic是一个用于移除接头和低质量数据的工具。[阅读更多]
使用Nextera文库制备试剂盒从NextSeq平台收集 Illumina 读数。
mkdir illumina |
预期输出:
illumina/NXT20x_R1_paired.fastq.gz |
数据质量控制bbmap
bbmap是一套用于测序读数质量控制的工具。它可用于删除冗余数据和 PhiX 对照。[阅读更多]
clumpify.sh是一个删除冗余数据的工具。[阅读更多]
bbduk.sh是一种去除污染数据的工具(例如,宿主基因组、PhiX 对照)。[阅读更多]
#clumpify |
预期输出:
illumina/NXT20x_R1_paired_dedup.fastq.gz |
基因组组装spades
spades是一个用于短读长的基因组组装器。[阅读更多]
spades.py --isolate -t 4 -1 illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz -2 illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz -o illumina/spades |
预计组装:
illumina/spades/contigs.fasta |
Q2:我们在这里从分离株中组装了细菌基因组。如果我们有宏基因组样本怎么办?
我们可以使用
--meta选项在spades中来组装宏基因组样本。[阅读更多]
使用 ONT 数据进行基因组组装
使用guppy(可选)或者porechop移除接头
本练习中未提供
guppy和porechop的安装如果您想使用它们,请尝试自行安装。
guppy是一种用于 ONT 数据的碱基识别和接头修剪的工具。guppy不是开源的,因此需要注册ONT帐户才能查看其文档和下载。[阅读更多]
porechop是一个开源工具,用于 ONT 数据的接头修剪。[阅读更多]
默认情况下,条形码将在guppy多路分解步骤中被修剪。我们不会对我们的数据重复进行条形码修剪。
如果您想修剪条形码,可以使用以下命令guppy。
guppy_barcoder -i data/wgs/ont_r10_20x.fastq.gz -s ont_r10/ont_r10_20x_barcoded.fastq.gz --barcode_kits EXP-NBD104 --trim_barcodes |
以及以下命令porechop。
porechop -i data/wgs/ont_r10_20x.fastq.gz -o ont_r10/ont_r10_20x_porechop.fastq.gz --threads 4 |
seqkit读取质量控制
seqkit是一种用于操作测序数据的工具。[阅读更多]这里我们用来seqkit去除短读和低质量数据。
mkdir ont_r10 |
使用seqkit stat来检查质量控制之前和之后的 ONT 读数。
seqkit stat data/wgs/ont_r10_20x.fastq.gz ont_r10/ont_r10_20x_f.fastq.gz |
预期输出:
file format type num_seqs sum_len min_len avg_len max_len |
flye基因组组装
flye是 ONT 推荐的长读基因组组装器。[阅读更多]
flye --nano-raw ont_r10/ont_r10_20x_f.fastq.gz --out-dir ont_r10/flye --threads 4 |
预期的组装输出:
ont_r10/flye/assembly.fasta |
Q3:默认
flye用于组装基因组。如果我们有宏基因组样本怎么办?我们可以使用
--meta选项来在flye中组装宏基因组样本。[阅读更多]
使用racon与medaka进行基因组矫正
长读长基因组组装程序通常会产生连续性高但准确性低的基因组草图。需要额外的矫正步骤来提高基因组草图的准确性。但最佳的矫正策略和工具仍在争论中。
racon是一种基于图的相似度算法,用于完善长读长基因组组装。[阅读更多]。
medaka是官方为ONT数据打造的基于神经网络的数据矫正工具。[阅读更多]
最近,ONT 推荐用medaka直接矫正flye组装后的数据。但结合racon和medaka仍然是一种常见的做法。
这里我们以medaka在例子:
由于medaka是基于神经网络的,选择合适的模型会影响抛光效果。您可以用来medaka tools list_models列出所有可用的模型。对于我们的示例,ONT 读数是从R10.4.1 flowcell收集的,并且guppy采用hac模式来做碱基生成。
medaka tools list_models |
预期结果:
ont_r10/medaka/consensus.fasta |
racon和medaka混合模式
mkdir ont_r10/racon |
预期的结果:
ont_r10/racon/racon.fasta |
ONT 和 Illumina 读取的混合组装
混合组装是结合不同测序技术优势的常见做法。这里我们选择两种常用的混合组装策略:
pilon:直接使用 ONT 组装作为主干,并用 Illumina 读取对其进行矫正。unicycler:短读优先混合组装。
pilonILLUMINA 读取矫正
pilon是一种用短读长优化基因组组装的工具。[阅读更多]
mkdir -p hybrid/pilon |
预期产出:
hybrid/pilon/pilon.fasta |
可选:unicycler混合组装
unicycler可以进行短读优先混合组装。[阅读更多]
这需要一些时间,因此我们将在示例中跳过这一步。完成其他步骤后,请随时返回此步骤。
Linux用户
unicycler -l ont_r10/ont_r10_20x_f.fastq.gz -1 illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz -2 illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz -o hybrid/unicycler --threads 4 |
MacOS 用户
由于on的最新MacOS版本落后于0.5.0,我们附加两个标志和来保持最大的兼容性。unicyclerconda--no_correct--no_pilon
unicycler --no_correct --no_pilon -l ont_r10/ont_r10_20x_f.fastq.gz -1 illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz -2 illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz -o hybrid/unicycler |
预期的产出:
hybrid/unicycler/assembly.fasta |
使用quast对组装基因组进行质量评估
我们已经生成了许多具有不同策略的程序集。让我们使用基于PacBio深度测序的完整组装作为参考基因组来评估每个数据集的质量。如果您尚未完成所有组装步骤,您可以使用./data/wgs/assemblies提供的组装文件。
seqkit stat data/wgs/assemblies/*.fasta data/wgs/ncbi_pacbio_TL110.fasta |
预期输出:
file format type num_seqs sum_len min_len avg_len max_len |
与参考相比,各个组装的总长度相似。
但序列数 (
num_seqs) 和最大序列长度 (max_len) 变化很大。
让我们用quast来检查各个组装的质量。
quast data/wgs/assemblies/*.fasta -r data/wgs/ncbi_pacbio_TL110.fasta -o quast |
您可以打开quast目录中的report.html文件来查看各个组装数据集的质量。
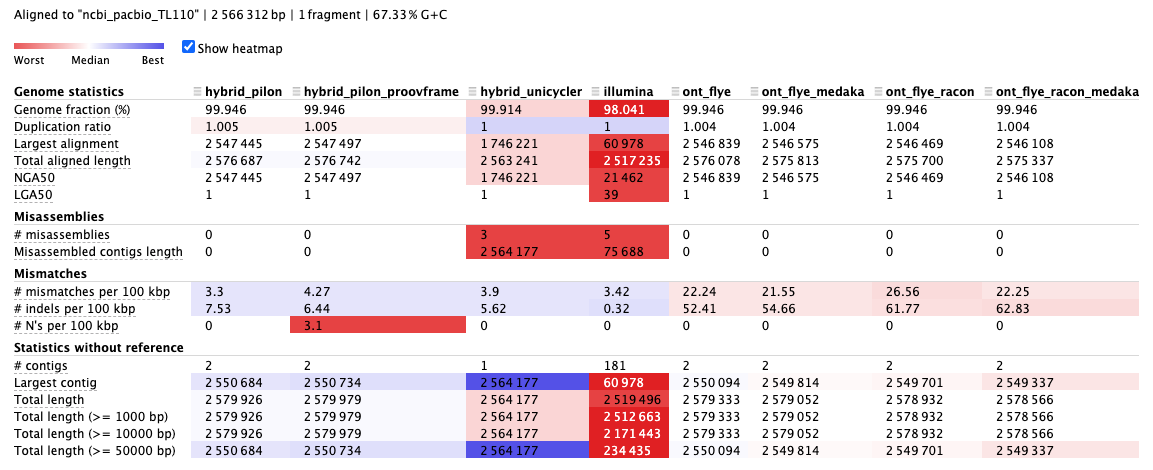
Q4:根据示例,您认为哪种测序技术在基因组完整性和连续性方面效果更好?
检查
Genome fraction和NGA50Q5:根据示例,您认为哪种装配的精度最好?
检查
Misassemblies和MismatchesQ6:根据示例,ONT 组件中的主要错误类型是什么?
检查
mismatches和indels。[阅读更多]Q7:您认为此示例的最佳组装策略是什么?
可选:参考引导校正proovframe
高频率indels会导致 ONT 组装的编码序列 (CDS) 发生移码。通过参考引导校正,我们可以校正 ONT 组装的 CDS 中的移码。
此步骤是可选的,如果您没有时间,可以跳过它。
为了使用该工具,由于依赖冲突,proovframe我们需要创建另一个conda环境。
conda deactivate |
基因组注释prokka
prokka是快速注释细菌基因组的常用工具。[阅读更多]
mkdir proovframe |
预期输出:
proovframe/prokka/pacbio.err |
我们有许多来自prokka的输出文件。这里我们仅使用翻译后的蛋白质序列(./proovframe/prokka/pacbio.faa文件)
移码校正proovframe
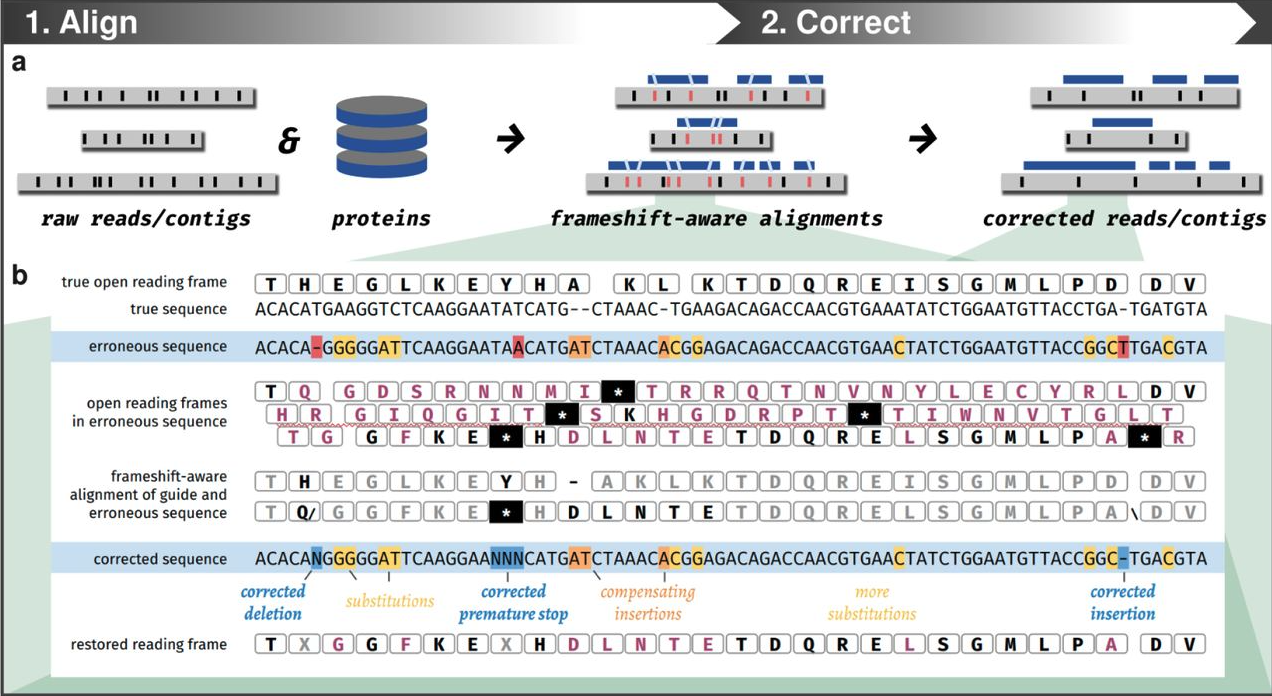
proovframe是 CDS 中基于参考序列的移码校正工具。[阅读更多]
proovframe map -a proovframe/prokka/pacbio.faa -o proovframe/pilon.tsv hybrid/pilon/pilon.fasta |
预期的输出:
proovframe/pilon_corrected.fasta |
Q8:在之前的报告中,
shybrid_pilon_proovframe.fasta中的N'会增加。你觉得这是好是坏?
本文为学习记录,课程作者为:YanHui
